
Strona Główna

PRECYZJA DZIAŁANIA
= BEZPRECEDENSOWE WYNIKI

v#Wytyczne NCCN dotyczące NDRP zawierają rekomendacje dotyczące niektórych pojedynczych biomarkerów, które powinny być testowane, i zalecają techniki testowania, ale nie wskazują żadnych konkretnych dostępnych na rynku testów biomarkerów ani laboratoriów komercyjnych6.
**Ozymertynib jest zalecany pacjentom z NDRP z mutacją EGFR (delecja w eksonie 19, substytucja w egzonie 21 L858R) w stadium zaawansowania IB-IIIA po całkowitej resekcji guza, którzy otrzymywali wcześniej chemioterapię uzupełniającą lub nie kwalifikują się do chemioterapii opartej na pochodnych platyny4.
††Szczegółowe rekomendacje, w tym inne opcje leczenia, znajdują się w wytycznych NCCN4.
NCCN=National Comprehensive Cancer Network®; (NCCN®;)


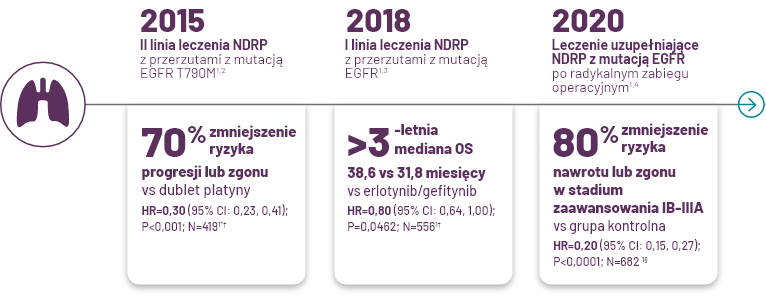
*W 2015 r. lek TAGRISSO®; został dopuszczony do stosowania jako terapia w 2 linii leczenia NDRP w IV stadium zaawansowania z mutacją T790M EGFR w procedurze przyspieszonej; na podstawie danych skumulowanych z badań klinicznych AURA faza „extension” (faza II) oraz AURA2. Pierwszorzędowy punkt końcowy ORR wynosił 59% (95% CI: 54, 64). Pełne dopuszczenie do obrotu zostało udzielone w 2017 r. na podstawie danych badania fazy III AURA32,5.
†Pierwszorzędowy punkt końcowy w badaniu AURA3. Mediana PFS wynosiła 10,1 miesiąca (95% CI: 8.3, 12.3) dla TAGRISSO®; w porównaniu z 4,4 miesiąca (95% CI: 4.2, 5.6) dla chemioterapii z wykorzystaniem pochodnych platyny1.
‡Drugorzędowy punkt końcowy w badaniu FLAURA. Pierwszorzędowym punktem końcowym był PFS: Mediana PFS - 18,9 miesiąca (95% CI: 15,2, 21,4) dla TAGRISSO®; w porównaniu z medianą PFS wynoszącą 10,2 miesiąca (95% CI: 9,6, 11,1) dla erlotynibu/gefitynibu (HR = 0,46 [95% CI: 0,37, 0,57]; P <0,0001)1.
§Drugorzędowy punkt końcowy w badaniu ADAURA. Mediana DFS nie została osiągnięta (95% CI: NE, NE) dla TAGRISSO®; i wynosiła 27,5 miesiąca (95% CI: 22,0, 35,0) dla grupy kontrolnej. Pierwszorzędowym punktem końcowym był DFS u pacjentów w stadium zaawansowania II/IIIA (HR = 0,17 [95% CI: 0,12, 0,23]; P <0,0001). Mediana DFS nie została osiągnięta dla TAGRISSO®; (95% CI: 38,8, NE) i wynosiła 19,6 miesiąca (95% CI: 16,6, 24,5) dla grupy kontrolnej. Dopuszczenie do obrotu na podstawie porównania z grupą kontrolną (placebo)1.
||Grupa kontrolna=placebo.
- Charakterystyka Produktu Leczniczego Tagrisso®; [dostęp: 09.2024]
- AstraZeneca Media Centre. AURA komunikat prasowy. https://www.astrazeneca.com/media-centre/press-releases/2015/TAGRISSO-AZD9291-approved-by-the-US-FDA-for-patients-with-EGFR-T790M-mutation-positive-metastatic-non-small-cell-lung-cancer-13112015.html. opublikowano 13 listopada , 2015 [dostęp: sierpień 2021].
- Odniesienie za zgodą NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®;) w przypadku niedrobnokomórkowego raka płuca V.3.2023. © National Comprehensive Cancer Network, Inc. 2023. Wszelkie prawa zastrzeżone[ Dostęp: 13 kwiecień 2023]. Aby zapoznać się z najnowszą i pełną wersją wytycznych, przejdź do NCCN.org. NCCN nie udziela żadnych gwarancji dotyczących ich treści, wykorzystania lub zastosowania i zrzeka się wszelkiej odpowiedzialności za ich zastosowanie lub wykorzystanie w jakikolwiek sposób.
- AstraZeneca Media Centre. AURA3 komunikat prasowy https://www.astrazeneca.com/media-centre/press-releases/2017/tagrisso-osimertinib-receives-us-fda-full-approval-31032017.html. Opublikowano 31 marzec, 2017. [Dostęp: 10 wrzesień, 2020.
WSKAZANIA

Należy zapoznać się w całości z Charakterystyką Produktu Leczniczego TAGRISSO®, w tym z ulotką dla pacjenta.
Może Pan/Pani zgłosić działania niepożądane związane z produktami firmy AstraZeneca klikając tutaj.
CI, przedział ufności; DFS, przeżycie wolne od choroby; EGFR, receptor naskórkowego czynnika wzrostu; EGFRm, mutacja receptora naskórkowego czynnika wzrostu; HR, współczynnik ryzyka; NE, niemożliwe do oszacowania; NDRP, niedrobnokomórkowy rak płuca; OS, przeżycie całkowite; PFS, czas przeżycia wolny od progresji choroby.
- Tsuboi M, Wu YL, Grohe C, i in. Osimertinib as adjuvant therapy in patients with resected EGFRm stage IB–IIIA NSCLC: updated results from ADAURA. Przedstawiono podczas: Kongres ESMO 2022; 9–13 września 2022 r.; Paryż, Francja.
- Charakterystyka Produktu Leczniczego Tagrisso® [dostęp: 02.2024]
PL-16691
Ocena mutacji w genie kodującym EGFR
Jeżeli rozważane jest zastosowanie produktu leczniczego TAGRISSO w leczeniu uzupełniającym po radykalnej resekcji guza u pacjentów z NDRP, potwierdzenie mutacji (delecja w egzonie 19 – Ex19del lub substytucji w egzonie 21 L858R) jest warunkiem koniecznym do inicjacji terapii ozymertynibem. Ocena obecności mutacji powinna zostać przeprowadzona w laboratoriach klinicznych z wykorzystaniem zwalidowanych testów na podstawie DNA z materiału histologicznego pozyskanego podczas biopsji lub z materiału pooperacyjnego. Jeżeli rozważane jest zastosowanie produktu leczniczego TAGRISSO w leczeniu miejscowo zaawansowanego lub uogólnionego NDRP, ważne jest, aby potwierdzona została obecność mutacji w genie kodującym EGFR. Oznaczenie należy wykonać przy użyciu walidowanej metody testowej z użyciem DNA pozyskanego z tkanki guza lub wolnego krążącego DNA nowotworowego (ctDNA) pozyskanego z osocza. Dodatni wynik oznaczenia statusu mutacji w genie kodującym EGFR (mutacje aktywujące w genie EGFR w przypadku leczenia pierwszego rzutu, mutacje delecji w eksonie 19 lub substytucji w eksonie 21 (L858R), gdy produkt leczniczy TAGRISSO jest podawany w skojarzeniu z pemetreksedem i chemioterapią opartą na pochodnych platyny w leczeniu pierwszego rzutu lub mutacje oporności T790M w przypadku progresji w trakcie lub po zakończonej terapii inhibitorami kinaz tyrozynowych) testem przeznaczonym do badania materiału z tkanki guza lub z próbki osocza wskazuje na to, że pacjent kwalifikuje się do leczenia produktem leczniczym TAGRISSO. Jednakże, w przypadku oznaczania mutacji na podstawie badania ctDNA z próbki osocza i uzyskania wyniku ujemnego zalecane jest, o ile tylko jest to możliwe, wykonanie testu z wykorzystaniem tkanki guza, ze względu na możliwość uzyskiwania wyników fałszywie ujemnych w badaniu z próbki osocza. Do oceny należy wykorzystać wyłącznie stabilne, wiarygodne i czułe testy o udowodnionej użyteczności w diagnostyce mutacji w genie kodującym EGFR. Śródmiąższowa choroba płuc (ang. ILD) U pacjentów leczonych produktem leczniczym TAGRISSO w ramach badań klinicznych obserwowano występowanie ciężkiej, zagrażającej życiu lub prowadzącej do zgonu śródmiąższowej choroby płuc (ang. Interstitial Lung Disease, ILD) lub reakcje podobne do ILD (np. zapalenie płuc). W większości przypadków poprawa lub całkowite ustąpienie tego stanu następowało po przerwaniu stosowania leku. Pacjenci, u których uprzednio występowała śródmiąższowa choroba płuc, lub lekopochodna ILD, lub popromienne zapalenie płuc wymagające leczenia steroidami oraz pacjenci z jakimikolwiek objawami klinicznie czynnej ILD byli wykluczani z udziału w badaniach klinicznych (patrz punkt 4.8 w ChPL). W badaniach klinicznych ADAURA, FLAURA, FLAURA2 i AURA występowanie śródmiąższowej choroby płuc (ILD) lub reakcji podobnych do ILD zgłaszano u 4,0% spośród 1813 pacjentów, którzy otrzymywali produkt leczniczy TAGRISSO w monoterapii. Odnotowano 7 zgonów w przypadku leczenia pacjentów z miejscowo zawansowanym i uogólnionym NDRP. Nie odnotowano żadnego zgonu w przypadku leczenia uzupełniającego ozymertynibem. Częstość występowania ILD wynosiła 11,2% wśród Japończyków, 2,3% wśród Azjatów niebędących Japończykami oraz 2,7% wśród pacjentów pochodzenia nieazjatyckiego (patrz punkt 4.8 w ChPL). W badaniu klinicznym FLAURA2 występowanie ILD lub reakcji podobnych do ILD zgłoszono u 3,3%, a przypadki śmiertelne u 0,4% (n=1) spośród 276 pacjentów, którzy otrzymywali produkt leczniczy TAGRISSO w skojarzeniu z pemetreksedem i chemioterapią opartą na pochodnych platyny. Częstość występowania ILD wynosiła 14,9% wśród Japończyków oraz 1,7% wśród pacjentów pochodzenia nieazjatyckiego; u żadnego spośród pacjentów pochodzenia azjatyckiego niebędących Japończykami nie wystąpiło zdarzenie ILD w grupie otrzymującej leczenie skojarzone w badaniu FLAURA2. Mediana czasu od podania pierwszej dawki do początku ILD lub reakcji podobnych do ILD wyniosła 161 dni. U wszystkich pacjentów, u których dojdzie do wystąpienia ostrych objawów i (lub) niewyjaśnionego nasilenia objawów ze strony układu oddechowego (np. duszności, kaszlu, gorączki), należy natychmiast wykonać badania w celu wykluczenia ILD. W trakcie tej diagnostyki należy wstrzymać stosowanie tego produktu leczniczego. W przypadku potwierdzenia rozpoznania śródmiąższowej choroby płuc należy zaprzestać stosowania produktu leczniczego TAGRISSO i wdrożyć odpowiednie postępowanie u pacjenta. Wznowienie podawania produktu leczniczego TAGRISSO należy rozważyć wyłącznie po dokładnym uwzględnieniu korzyści i ryzyka u danego pacjenta. Ciężkie skórne działania niepożądane (ang. severe cutaneous adverse reactions, SCARs) W związku z leczeniem produktem leczniczym TAGRISSO raportowano przypadki zespołu Stevensa-Johnsona (SJS) i toksycznej nekrolizy naskórka (TEN) z częstością występowania mieszczącą się odpowiednio w kategorii „rzadko” i „częstość nieznana”. Przed rozpoczęciem leczenia należy pouczyć pacjentów o przedmiotowych i podmiotowych objawach SJS i TEN. W razie wystąpienia objawów przedmiotowych i podmiotowych sugerujących SJS lub TEN, leczenie produktem TAGRISSO należy przerwać. W przypadku rozpoznania SJS lub TEN leczenie produktem TAGRISSO należy natychmiast zakończyć. Wydłużenie odstępu QTc U pacjentów leczonych produktem leczniczym TAGRISSO może wystąpić wydłużenie odstępu QTc. Wydłużenie odstępu QTc może prowadzić do zwiększenia ryzyka wystąpienia tachyarytmii komorowych (np. torsade de pointes) lub nagłego zgonu. W badaniach ADAURA, FLAURA, FLAURA2 i AURA nie zgłaszano występowania u pacjentów jakichkolwiek arytmii związanych z wydłużeniem odstępu QTc (patrz punkt 4.8 w ChPL). Pacjenci z klinicznie istotnymi zaburzeniami rytmu i przewodzenia stwierdzonymi na podstawie spoczynkowych zapisów elektrokardiograficznych (EKG) (np. z odstępem QTc powyżej 470 msec) byli wykluczeni z udziału w tych badaniach (patrz punkt 4.8 w ChPL). O ile to możliwe, należy unikać stosowania produktu leczniczego TAGRISSO u pacjentów z wrodzonym zespołem wydłużonego odstępu QT. Należy rozważyć okresowe monitorowanie z wykonywaniem zapisów elektrokardiograficznych (EKG) oraz oznaczeń stężeń elektrolitów u pacjentów z zastoinową niewydolnością serca, zaburzeniami gospodarki elektrolitowej, a także u tych pacjentów, którzy przyjmują produkty lecznicze, o których wiadomo, że wydłużają odstęp QTc. Należy wstrzymać stosowanie u pacjentów, u których dojdzie do wydłużenia odstępu QTc powyżej 500 ms w co najmniej 2 odrębnych badaniach EKG do czasu powrotu odstępu QTc do wartości mniejszej niż 481 ms lub do powrotu odstępu QTc do wartości wyjściowej, jeżeli odstęp QTc wynosi 481 ms lub więcej, a następnie należy wznowić stosowanie produktu leczniczego TAGRISSO w zmniejszonej dawce zgodnie z instrukcjami podanymi w Tabeli 1. Należy trwale zaprzestać stosowania produktu leczniczego TAGRISSO u pacjentów, u których nastąpi wydłużenie odstępu QTc w połączeniu z którymkolwiek spośród następujących zaburzeń: torsade de pointes, polimorficzny częstoskurcz komorowy, objawy ciężkich zaburzeń rytmu serca. Zmiany kurczliwości serca W badaniach klinicznych, u pacjentów leczonych lekiem TAGRISSO w monoterapii, u których wykonano pomiar frakcji wyrzutowej lewej komory serca (ang. LVEF) w warunkach wyjściowych oraz co najmniej jeden pomiar kontrolny, zmniejszenie LVEF o 10 punktów procentowych lub większe oraz zmniejszenie do mniej niż 50% występowały u 4,2% pacjentów (65/1557). U pacjentów z kardiologicznymi czynnikami ryzyka oraz z zaburzeniami, które mogą wpływać na LVEF, należy rozważyć prowadzenie monitorowania czynności serca, w tym pomiary LVEF w warunkach wyjściowych oraz w trakcie leczenia. U pacjentów, u których wystąpią istotne objawy przedmiotowe lub podmiotowe ze strony serca podczas leczenia, należy rozważyć prowadzenie monitorowania czynności serca, w tym pomiary LVEF. W przypadku kontrolowanego placebo badania ADAURA z zastosowaniem produktu leczniczego TAGRISSO w leczeniu uzupełniającym 1,5% (5/325) w grupie pacjentów otrzymujących ozymertynib oraz 1,5% (5/331) z grupy kontrolnej doświadczyło LVEF zmniejszenie frakcji wyrzutowej lewej komory (LVEF) o 10 punktów procentowych lub większe oraz zmniejszenie do mniej niż 50%. W badaniu FLAURA2 u 8,0% (21/262) pacjentów leczonych produktem TAGRISSO w skojarzeniu z pemetreksedem i chemioterapią opartą na pochodnych platyny, u których wykonano pomiar LVEF w warunkach wyjściowych oraz co najmniej jeden pomiar kontrolny wystąpiło zmniejszenie LVEF o 10 punktów procentowych lub większe oraz zmniejszenie do mniej niż 50%. Zapalenie rogówki zgłaszano u 0,6% (n=10) spośród 1813 pacjentów leczonych lekiem TAGRISSO w monoterapii w ramach badań ADAURA, FLAURA, FLAURA2 i AURA. Pacjenci z objawami przedmiotowymi i podmiotowymi wskazującymi na zapalenie rogówki, takimi jak ostre lub nasilające się: zapalenie oka, łzawienie, nadwrażliwość na światło, niewyraźne widzenie, ból oka i (lub) zaczerwienienie oka, powinni zostać niezwłocznie skierowani do lekarza okulisty (patrz punkt 4.2 Tabela 1 w ChPL). Niedokrwistość aplastyczna W związku z leczeniem produktem leczniczym TAGRISSO zgłaszano rzadkie przypadki niedokrwistości aplastycznej, w tym zdarzenia śmiertelne. Przed rozpoczęciem leczenia należy pacjentów pouczyć w zakresie przedmiotowych i podmiotowych objawów niedokrwistości aplastycznej, w tym między innymi utrzymującej się gorączki, powstawania wylewów podskórnych, krwawienia, bladości, zakażenia i uczucia zmęczenia. Jeśli wystąpią przedmiotowe i podmiotowe objawy sugerujące niedokrwistość aplastyczną, należy rozważyć ścisłe monitorowanie pacjenta i przerwanie lub zakończenie podawania produktu leczniczego TAGRISSO. Leczenie produktem TAGRISSO należy zakończyć u pacjentów z potwierdzoną niedokrwistością aplastyczną (patrz punkt 4.2 w ChPL). Wiek i masa ciała Pacjenci w podeszłym wieku (> 65 lat) lub pacjenci o niskiej masie ciała (<50 kg) mogą być narażeni na zwiększone ryzyko wystąpienia działań niepożądanych stopnia 3. lub wyższego. Zaleca się ścisłe monitorowanie tych pacjentów (patrz punkt 4.8 w ChPL). Sód Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) w tabletce, więc zasadniczo jest on „wolny od sodu”