
NIE CZEKAJ – LECZ SKUTECZNIE OD POCZĄTKU

Nowy standard leczenia HER2-dodatniego zaawansowanego raka piersi1-3
Mechanizm działania
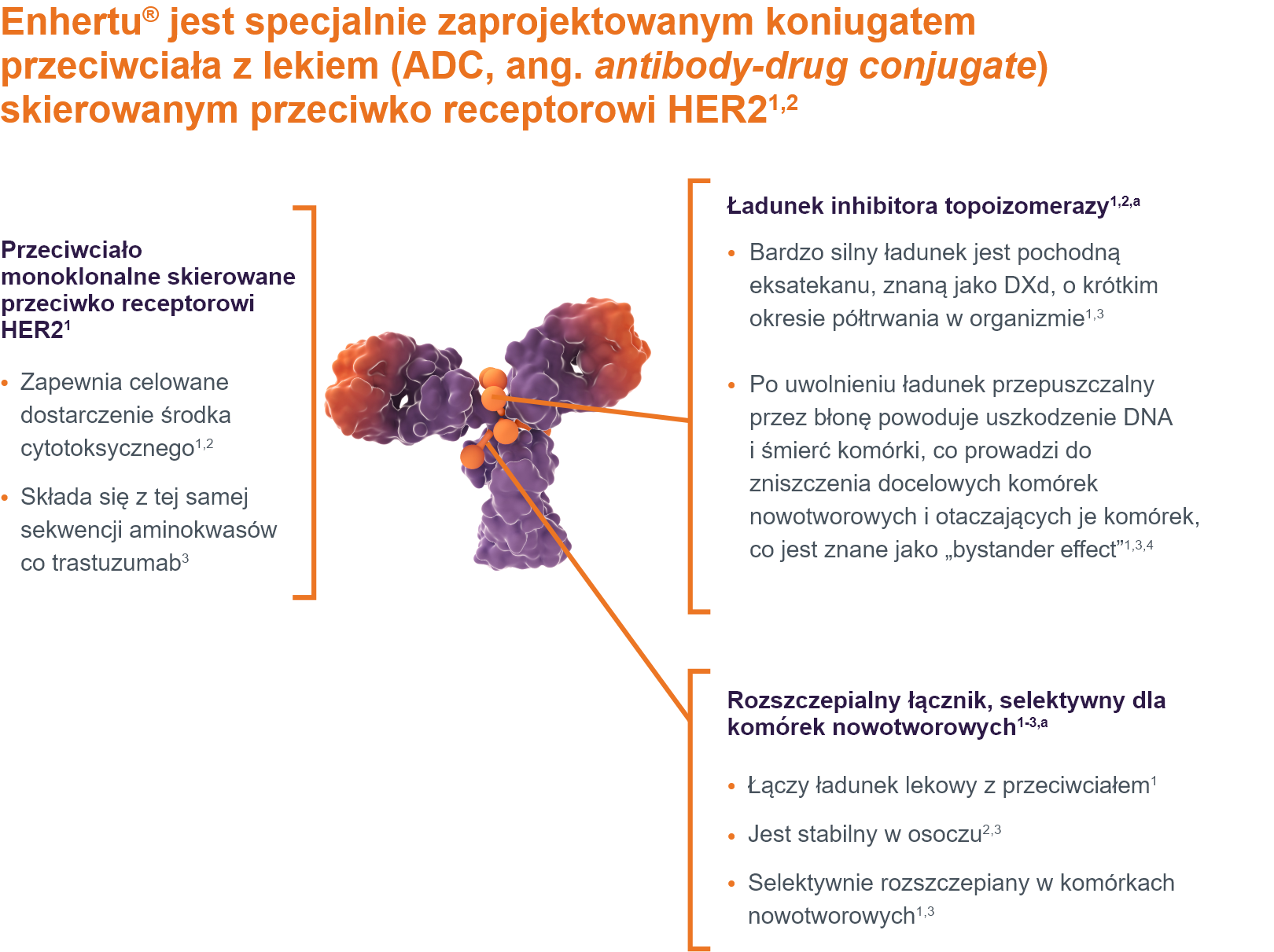
-
a. W oparciu o niekliniczne badania in vitro i in vivo. Znaczenie kliniczne tych cech jest obecnie badane.
-
b. ADC to mieszanina cząsteczek, w których DAR jest zmienny. Jednorodność DAR odnosi się do mieszaniny, w której występuje niska zmienność DAR; liczba ładunku na przeciwciało mieści się w wąskim zakresie.
-
1. Iwata H. i wsp. Zaprezentowano na: Coroczne spotkanie ASCO; 1–5 czerwca 2018; Chicago, IL. Abstrakt 2501; https://ascopubs.org/doi 10.1200/JCO.2018.36.15_suppl.2501
-
2. Nakada T. i wsp. Chem Pharm Bull (Tokio). 2019;67(3):173–185;
-
3. Krop I. i wsp. Zaprezentowano na: SABCS 2019, 10–14 grudnia. San Antonio, USA. Abstrakt #GS1-03;
-
4. Aktualna charakterystyka produktu leczniczego ENHERTU.
Dawkowanie,
przygotowanie i podawanie
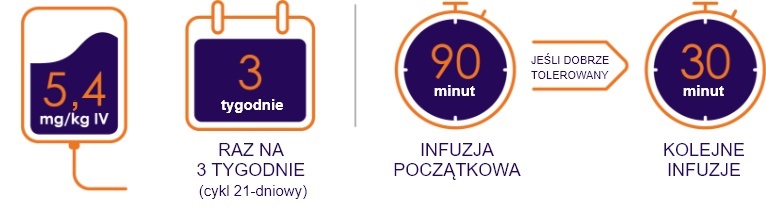
Zalecany schemat oraz dawkowanie zależne od masy ciała1
-
Produkt leczniczy Enhertu® zawsze podaje się w monoterapii
-
Dawka Enhertu® w mBC (5,4 mg/kg) może różnić się od innych zatwierdzonych wskazań
Zalecane zmniejszenie dawki produktu leczniczego Enhertu® w przypadku działań niepożądanych1
-
Postępowanie w przypadku działań niepożądanych może wymagać czasowego przerwania leczenia, zmniejszenia dawki lub zaprzestania leczenia produktem leczniczym Enhertu® zgodnie z modyfikacjami dawki podanymi w poniższych tabelach
-
Nie należy ponownie zwiększać dawki produktu leczniczego Enhertu® po jej zmniejszeniu.
-
W przypadku opóźnienia lub pominięcia planowanej dawki, należy ją podać jak najszybciej; nie należy czekać do następnego planowanego cyklu. Należy dostosować schemat podawania, aby zachować 3-tygodniowy odstęp między dawkami. Podawać infuzję w dawce i szybkości tolerowanej przez pacjenta w ostatniej infuzji.
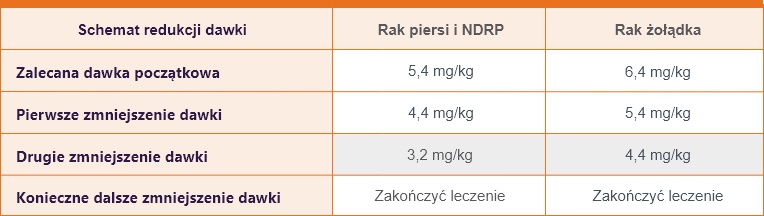
Modyfikacja dawki w przypadku działań niepożądanych1
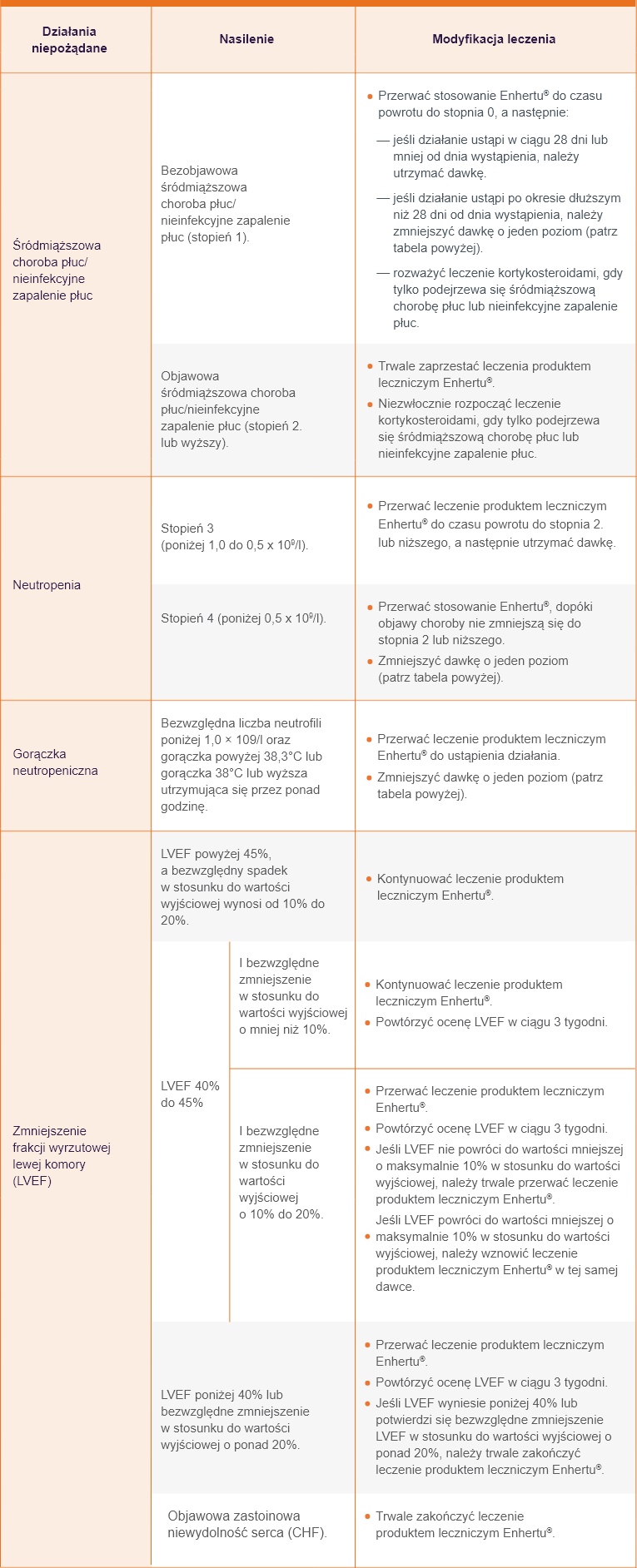
Stopnie toksyczności są zgodne z NCI-CTCAE v 5.0.
Przygotowanie produktu leczniczego Enhertu® do podawania
Aby zapobiec błędom w stosowaniu leku, należy sprawdzić etykiety fiolki, aby upewnić się, że lekiem przygotowywanym i podawanym jest Enhertu®, a nie trastuzumab lub trastuzumab emtanzyna.1
Przed infuzją dożylną należy zrekonstytuować i dodatkowo rozcieńczyć produkt leczniczy Enhertu®. Stosować odpowiednią technikę aseptyczną.1
Należy przestrzegać obowiązujących specjalnych procedur postępowania i utylizacji.1
Rekonstytucja1

-
Zrekonstytuować bezpośrednio przed rozcieńczeniem.
-
Do podania pełnej dawki konieczne może być użycie więcej niż jednej fiolki. Należy obliczyć dawkę (mg), całkowitą wymaganą objętość rozpuszczonego roztworu oraz liczbę potrzebnych fiolek Enhertu®.
-
Zawartość każdej 100 mg fiolki należy zrekonstytuować przy użyciu jałowej strzykawki, powoli wstrzykując 5 ml wody do wstrzykiwań do każdej fiolki, aby uzyskać końcowe stężenie 20 mg/ml.
-
Delikatnie obracać fiolką, aż do całkowitego rozpuszczenia. Nie wstrząsać.
-
Z mikrobiologicznego punktu widzenia produkt leczniczy należy zużyć natychmiast. Wykazano, że produkt, który nie zostanie zużyty natychmiast, zachowuje stabilność chemiczną i fizyczną w trakcie użytkowania do 48 godzin w temperaturze od 2 ºC do 8 ºC. Fiolki z przygotowanym roztworem produktu leczniczego Enhertu należy przechowywać w lodówce w temperaturze od 2 °C do 8 °C, chroniąc przed światłem. Nie zamrażać.
-
Przygotowany do podania produkt leczniczy nie zawiera środków konserwujących i jest przeznaczony wyłącznie do jednorazowego użytku.
Rozcieńczenie1

-
Pobrać obliczoną objętość z fiolki (fiolek) przy użyciu jałowej strzykawki. Ocenić zrekonstytuowany roztwór wzrokowo pod kątem występowania cząstek stałych i zmiany barwy.
- Roztwór powinien być przejrzysty i bezbarwny do jasnożółtego. Nie stosować, jeśli widoczne są cząstki lub jeśli roztwór jest mętny albo przebarwiony.
-
Rozcieńczyć obliczoną objętość przygotowanego roztworu produktu leczniczego Enhertu w worku infuzyjnym zawierającym 100 ml 5% roztworu glukozy. Nie używać roztworu chlorku sodu. Zaleca się użycie worka infuzyjnego z polichlorku winylu lub poliolefiny (kopolimer etylenu i polipropylenu).
-
Delikatnie odwrócić worek infuzyjny, aby dokładnie wymieszać roztwór. Nie wstrząsać.
-
Zakryć worek infuzyjny w celu ochrony przed światłem.
-
Jeśli produkt leczniczy nie zostanie zużyty natychmiast, przechowywać w temperaturze pokojowej do 4 godzin, co obejmuje przygotowanie i podanie wlewu, lub w lodówce w temperaturze od 2°C do 8°C do 24 godzin, chroniąc przed światłem. Nie zamrażać.
-
Niewykorzystaną pozostałość produktu leczniczego w fiolce należy wyrzucić.
Podawanie wlewu1

-
Jeśli przygotowany roztwór do infuzji był przechowywany w lodówce (2ºC do 8ºC), zaleca się przed podaniem odczekać, aż osiągnie temperaturę pokojową, chroniąc go przed światłem.
-
Produkt leczniczy Enhertu® należy podawać we wlewie dożylnym wyłącznie z wbudowanym filtrem polieterosulfonowym (PES) lub polisulfonowym (PS) o średnicy 0,20 lub 0,22 mikrona.
-
Początkową dawkę należy podać w 90-minutowym wlewie dożylnym. Jeśli wcześniejszy wlew był dobrze tolerowany, kolejne dawki Enhertu® można podawać w postaci 30-minutowych wlewów. Nie podawać we wstrzyknięciu dożylnym ani w bolusie (patrz punkt 4.2).
-
Zakryć worek infuzyjny w celu ochrony przed światłem.
-
Jeśli produkt leczniczy nie zostanie zużyty natychmiast, fiolki z przygotowanym roztworem trastuzumabu derukstekanu należy przechowywać w lodówce w temperaturze od 2 °C do 8 °C do 24 godzin od momentu zrekonstytuowania, chroniąc przed światłem. Nie zamrażać
-
Nie mieszać produktu leczniczego Enhertu® z innymi produktami leczniczymi ani nie podawać innych produktów leczniczych przez tę samą linię dożylną.
- Aktualna charakterystyka produktu leczniczego Enhertu®.
Profil bezpieczeństwa
Śródmiąższowa choroba płuc/nieinfekcyjne zapalenie płuc1
Podczas stosowania produktu leczniczego Enhertu® zgłaszano przypadki śródmiąższowej choroby płuc i (lub) nieinfekcyjnego zapalenia płuc. Obserwowano przypadki zgonu.
Należy zalecić pacjentom natychmiastowe zgłaszanie kaszlu, duszności, gorączki i (lub) wszelkich nowych lub nasilających się objawów ze strony układu oddechowego. Pacjentów należy monitorować pod kątem objawów przedmiotowych i podmiotowych śródmiąższowej choroby płuc/nieinfekcyjnego zapalenia płuc. Oznaki śródmiąższowej choroby płuc/nieinfekcyjnego zapalenia płuc należy niezwłocznie zbadać. Pacjentów z podejrzeniem śródmiąższowej choroby płuc lub nieinfekcyjnego zapalenia płuc należy ocenić za pomocą obrazowania radiograficznego, najlepiej tomografii komputerowej (TK). Należy rozważyć konsultację z pulmonologiem. W przypadku bezobjawowej (stopnia 1.) śródmiąższowej choroby płuc lub bezobjawowego nieinfekcyjnego zapalenia płuc należy rozważyć leczenie kortykosteroidami (np. ≥ 0,5 mg/kg/dobę prednizolonu lub jego odpowiednika).
Podawanie produktu leczniczego Enhertu® należy wstrzymać do czasu powrotu do stopnia 0. i można je wznowić zgodnie z instrukcją w tabeli powyżej. W przypadku objawowej śródmiąższowej choroby płuc lub objawowego nieinfekcyjnego zapalenia płuc (stopnia 2. lub wyższego) należy niezwłocznie rozpocząć leczenie kortykosteroidami (np. ≥ 1 mg/kg/dobę prednizolonu lub jego odpowiednika) i kontynuować przez co najmniej 14 dni, a następnie stopniowo odstawiać przez co najmniej 4 tygodnie. Produkt leczniczy Enhertu® należy całkowicie odstawić u pacjentów, u których stwierdzono objawową (stopnia 2. lub wyższego) śródmiąższową chorobę płuc lub objawowe nieinfekcyjne zapalenie płuc. Pacjenci ze śródmiąższową chorobą płuc lub nieinfekcyjnym zapaleniem płuc w wywiadzie lub pacjenci z umiarkowanymi lub ciężkimi zaburzeniami czynności nerek mogą być narażeni na zwiększone ryzyko wystąpienia śródmiąższowej choroby płuc lub nieinfekcyjnego zapalenia płuc i powinni być starannie monitorowani.
Neutropenia1
W badaniach klinicznych produktu leczniczego Enhertu® zgłaszano przypadki neutropenii zakończone zgonem, w tym gorączki neutropenicznej. Przed rozpoczęciem stosowania produktu Enhertu® i przed podaniem każdej dawki oraz zgodnie ze wskazaniami klinicznymi należy wykonać pełną morfologię krwi. W oparciu o nasilenie neutropenii może być konieczne przerwanie podawania lub zmniejszenie dawki produktu leczniczego Enhertu®.
Zmniejszenie frakcji wyrzutowej lewej komory1
W trakcie stosowania terapii anty-HER2 obserwowano zmniejszenie frakcji wyrzutowej lewej komory (LVEF). Przed rozpoczęciem stosowania produktu leczniczego Enhertu® oraz w regularnych odstępach czasu podczas leczenia, zgodnie ze wskazaniami klinicznymi, należy wykonywać standardowe badania czynności serca (echokardiogram lub badanie MUGA [wielobramkowa angiografia radioizotopowa]) w celu oceny LVEF.
W przypadku zmniejszenia LVEF należy leczenie przerywać. W przypadku stwierdzenia LVEF poniżej 40% lub bezwzględnego zmniejszenia w stosunku do wartości wyjściowej o ponad 20% należy trwale odstawić produkt leczniczy Enhertu®. Produkt leczniczy Enhertu® należy trwale odstawić u pacjentów z objawową zastoinową niewydolnością serca (CHF) (patrz tabela powyżej).
Toksyczne działanie na zarodek i płód1
Produkt leczniczy Enhertu® może spowodować uszkodzenie płodu, jeśli zostanie podany kobiecie w ciąży. W doniesieniach po wprowadzeniu produktu leczniczego do obrotu stosowanie trastuzumabu, antagonisty receptora HER2, w okresie ciąży powodowało przypadki małowodzia, objawiające się śmiertelną hipoplazją płuc, nieprawidłowościami kostnymi i zgonem noworodka. Na podstawie wyników badań na zwierzętach oraz mechanizmu działania, inhibitor topoizomerazy I, składnik produktu leczniczego Enhertu®, DXd, może również powodować uszkodzenie zarodka i płodu, gdy jest podawany kobiecie w ciąży.
U kobiet w wieku rozrodczym należy przed rozpoczęciem stosowania produktu leczniczego Enhertu® sprawdzić, czy pacjentka jest w ciąży. Pacjentkę należy poinformować o potencjalnych zagrożeniach dla płodu. Kobietom w wieku rozrodczym należy doradzić stosowanie skutecznej metody antykoncepcji w trakcie leczenia i przez co najmniej 7 miesięcy po przyjęciu ostatniej dawki produktu leczniczego Enhertu®. Należy doradzić mężczyznom, których partnerki są w wieku rozrodczym, aby stosowali skuteczną metodę antykoncepcji w trakcie leczenia produktem leczniczym Enhertu® i przez co najmniej 4 miesiące po przyjęciu ostatniej dawki produktu leczniczego Enhertu®.
Pacjenci z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby1
Istnieją ograniczone dane dotyczące pacjentów z umiarkowanymi zaburzeniami czynności wątroby i brak danych dotyczących pacjentów z ciężkimi zaburzeniami czynności wątroby. Ponieważ metabolizm i wydalanie z żółcią są głównymi drogami eliminacji inhibitora topoizomerazy I, DXd, produkt leczniczy Enhertu® należy podawać ostrożnie pacjentom z umiarkowanymi i ciężkimi zaburzeniami czynności wątroby.
Najczęstsze (≥ 20%) zdarzenia niepożądane1
-
Nudności (75,0%), zmęczenie (57,3%), wymioty (42,1%), łysienie (37,6%), neutropenia (35,2%), zaparcia (35,0%), niedokrwistość (34,4%), zmniejszenie łaknienia (33,1%), biegunka (28,8%), zwiększenie aktywności aminotransferaz (26,5%), bóle mięśniowo-szkieletowe (26,2%), małopłytkowość (24,5%) i leukopenia (23,7%).
- Aktualna charakterystyka produktu leczniczego Enhertu®.
PL-20647